Síndrome de Rett 【Trastorno Neurológico Ligado al Cromosoma X】

¿Qué es el síndrome de Rett? Se trata de una enfermedad extraña, que ataca generalmente a las niñas y fue descubierta por el neurólogo Andrea Rett, a mediados de la década del 60 del siglo pasado.
Este médico detectó que más de 20 niñas presentaban un cuadro clínico similar, caracterizado por retardo mental, pérdida del lenguaje y otros síntomas producidos por un trastorno neurológico ligado al cromosoma X.
La enfermedad, en poco menos de 70 años, ha contado con investigaciones muy serias y casi un centenar de publicaciones referidas a las causas, tratamientos y avances científicos para curar la enfermedad.
El síndrome de Rett es una enfermedad poco conocida, no obstante, los avances médicos revelan que tienen las causas bien definidas y los tratamientos surten los efectos esperados para brindarles una mejor calidad de vida a las pacientes.
Se conoce de las investigaciones sobre el síndrome de Rett que, después de descubierta las expresiones clínicas, se precisó cuál era el gen que las provocaba y, en el año 2007, se comprueba en ratones que es posible restaurar el gen alterado.
¿Cuál es la Causa del Síndrome de Rett?

El trastorno está asociado a las mutaciones, aparentemente espontáneas, del gen conocido por las siglas MECP2, ligadas al cromosoma X.
El análisis de este gen llevó a la conclusión de que es una proteína vital para la función neural. De igual forma, este gen se asocia con otros genes en función activadora o de represión.
Además, es un mediador de la función epigénica, es decir, del comportamiento de la cadena genética, sin modificación en su secuencia en el ADN. Este gen es muy complejo por la cantidad de funciones que hace en la cadena genética y que varía según el tipo de células.
Algunos investigadores señalan que el gen causante del síndrome de Rett, afecta la forma y función de muchos genes responsables de la producción de proteínas que necesita el cerebro.
Visto así, se trata de un tramado complejo de funciones entre genes. Pero lo importante es que se tiene bien identificada la causa que desencadena el síndrome de Rett, aunque no es degenerativo, se ha demostrado que el cerebro sufre un cambio significativo de su tamaño original.
¿Cómo se Detecta el Síndrome de Rett?
Para detectar el síndrome de Rett hay que aplicar varias pruebas, porque se tiende a confundir con otras enfermedades.
Así que, lo ideal frente a las sospechas de que algo no está funcionando bien con tu niña, acude al especialista para que realicen algunos de estos diagnósticos:
- Pruebas sanguíneas: Para medir las proteínas en sangre, las enzimas hepáticas, los electrolitos y otros.
- Valoración del funcionamiento de la glándula tiroides.
- Análisis de cerebro: El diagnóstico del cerebro debe incluir un electroencefalograma, para observar la fisiología del cerebro. También es necesario analizar la conformación de músculos y nervios, así como la velocidad en la conducción nerviosa de los impulsos que existen entre órganos del cuerpo.
- Prueba genética: Esta prueba es la más importante porque arroja el comportamiento del ADN y las mutaciones del cromosoma X.
Síndrome de Rett en Varones
Los varones, aunque tienen una carga cromosómica distinta a las de las niñas, cuando sufren del síndrome de Rett, no sobreviven.
Las estadísticas señalan que fallecen antes de nacer o en los primeros meses de nacidos.
Cuáles son las Características del Síndrome de Rett

Los bebes que sufrirán del síndrome de Rett nacen con aspecto normal, e inclusive hasta el año y medio muestran un desarrollo acorde con su edad.
Sin embargo, después de esta etapa, comienzan a presentar los siguientes síntomas:
1- Desaceleración del Crecimiento
Los bebés con síndrome de Rett, después del año y medio de nacido, comienzan a presentar un menor crecimiento de su cabeza con respecto al cuerpo.
Junto a la microcefalia, otras partes del cuerpo también registran menores tamaños de lo normal.
2- Control Reducido del Movimiento
Entre los primeros signos de este síntoma destaca que el bebé no coordina bien sus movimientos y cada día que pasa los realiza de forma más lenta. Generalmente, la dificultad se aprecia en las manos y piernas.
Con el tiempo, los músculos se pueden tornar rígidos o débiles, causando que la persona asuma posiciones anormales cuando está sentada o de pie.
3- Pérdida del Habla y la Visión
Las niñas con este síndrome pierden la comunicación verbal definitivamente, mientras que el contacto visual sufre reveses, pero se puede volver a ganar con el tiempo.
En estas condiciones, generalmente, se pierde el interés por los juguetes y la socialización.
4- Movimientos Extraños
Los movimientos extraños los efectúan con sus manos y ojos. En el primer caso, generalmente, las estrujan, aplauden, las aprietan, se las lamen, o las frotan sin razón aparente.
Por otro lado, las niñas pueden ver de forma fija e intensa, parpadean constantemente, cruzan los ojos o cierran uno a la vez.
También pueden presentarse movimientos extraños como rechinar los dientes, reírse sin motivo, halarse el pelo, agarrarse la ropa, se irritan fácilmente rompiendo en llanto o gritos, sufren de miedos y ansiedad, etc.
¿Cuánto puede Vivir una Persona con Síndrome de Rett?
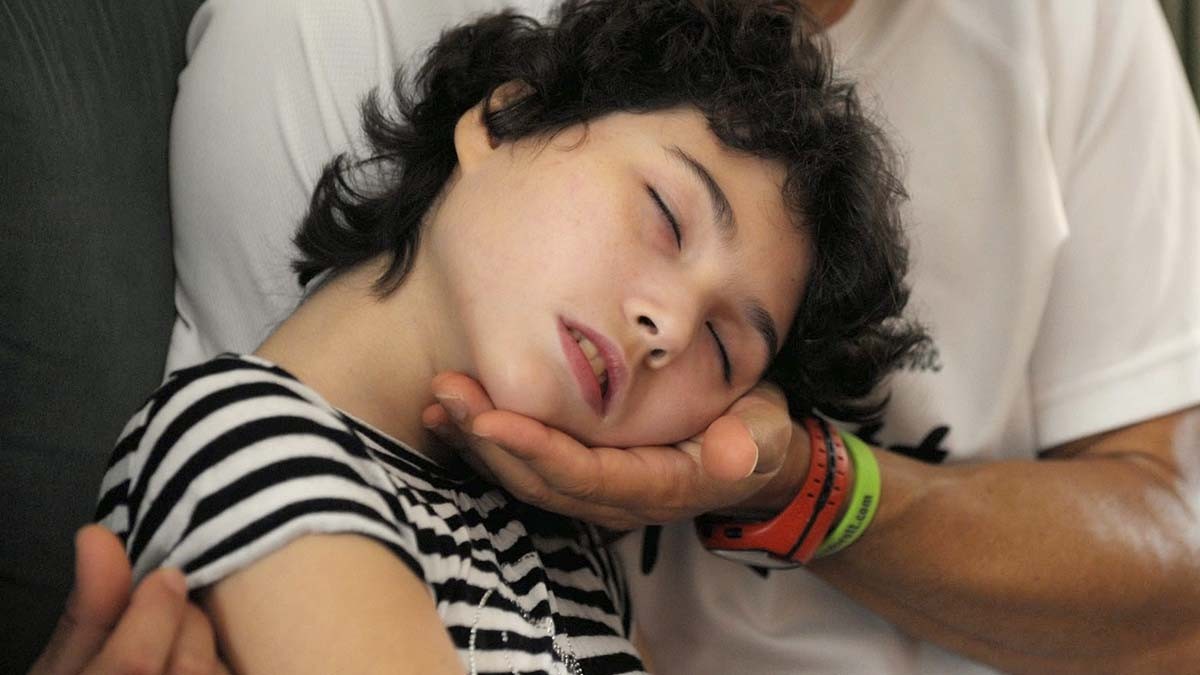
Las personas con síndrome de Rett pueden llegar a adultos, sin embargo, los factores de riesgos juegan en contra de la prolongación de la vida.
Durante los años que viven, padecen de mal nutrición porque sus músculos de la cara han sufrido deterioro y no les permiten masticar bien.
Padecen de problemas respiratorios y cardíacos. En la mayoría de los casos, el funcionamiento de la vejiga e intestinos no son los adecuados, generando incontinencias, estreñimientos y otros síntomas.
Etapas de Síndrome de Rett
El síndrome de Rett se desarrolla muy rápido, pues antes de los 10 años registra 4 etapas de desarrollo, en las que se deterioran rápidamente las funciones y el cuerpo de las niñas. Las 4 etapas del síndrome de Rett son:
1- En 18 meses
En esta primera etapa, las niñas pierden el contacto visual con su entorno, y le cuesta aprender a sentarse y a gatear.
2- Hasta los 4 años
Entre los 18 meses y 4 años, la niña muestra signos evidentes de disminución del crecimiento de la cabeza y comienza a realizar los movimientos extraños de mano y ojos.
De igual forma, se expresa con gritos, llantos, sufre de hiperventilación y deja de coordinar sus movimientos.
3- Entre los 4 y 10 años
Esta es una etapa crucial, debido a que se pueden controlar los movimientos y se mejoran los estados anímicos. Sin embargo, aparecen las convulsiones.
4- Después de los 10 años
La enfermedad ya está desarrollada totalmente, el paciente sufre escoliosis por el deterioro de la columna, se estabiliza la comunicación y algunas habilidades manuales, así como la frecuencia de las convulsiones.
Síndrome de Rett Tratamientos
El tratamiento para este síndrome es variado, pudiendo aplicarse terapias, cuidados alimentarios, ingesta de algunos medicamentos y los procedimientos quirúrgicos a los que haya lugar.
Las Terapias para el Síndrome de Rett
Suelen ser terapias del lenguaje y coordinación de movimientos corporales. También pueden incorporarse las actividades lúdicas.
- Tratamientos farmacológicos: Se procura controlar las convulsiones con la ingesta de antiepilépticos. Además, se logran controlar el estreñimiento, las alteraciones del sueño y los estados depresivos.
- Consumir alimentos sanos: La ingesta diaria debe ser alta en calorías y con suplementos vitamínicos como el calcio y la vitamina D, para mitigar los efectos de la osteoporosis.
- Quirúrgicas: Las pacientes pueden ser sometidas a cirugías para corregir los problemas de escoliosis o de reflujo gastroesofágico.