
¿Qué es la fenilcetonuria? La Fenilcetonuria está dentro del grupo de enfermedades raras, por su poca frecuencia.
También se le conoce como trastorno PKU, en el cual los pacientes no pueden descomponer el aminoácido llamado fenilalanina. Por tanto, este se acumula, generando graves consecuencias en la salud.
La fenilalanina, al no ser descompuestas, se convierte en una tóxica peligrosa para el cerebro, pero en condiciones normales, es un componente esencial en la formación de tejidos y células en el organismo.
Los pacientes con fenilcetonuria deben seguir una dieta estricta y reducida de proteínas, para evitar acelerar el deterioro del cerebro y otros órganos. Por eso, el diagnóstico temprano es muy importante.
Causas de la Fenilcetonuria
Las causas que originan la fenilcetonuria son de origen genético. Este trastorno se hereda de ambos padres. Toda persona tiene 42 cromosomas, 23 corresponden al padre y 23 a la madre.
Cuando uno de ellos tiene alterado el gen llamado PAH, no se produce la enfermedad en el bebé, pero este será portador del gen que mutó. Pero, si ambos padres se unen y tienen el gen alterado, trasmiten la enfermedad y esta se desarrolla.
Se ha estudiado que la incidencia de este trastorno es de una persona por cada diez mil. Añaden los investigadores que, depende también de la zona geográfica que se estudie, pero que, en términos generales, el 2% de la población total del mundo, padece de fenilcetonuria.
¿Por qué se Altera un Gen?
Los expertos en genética humana, manejan la tesis de que los genes se alteran, producto de "accidentes" en la cadena celular, y que todos los seres humanos tenemos, por lo menos, 10% de genes alterados en nuestros cromosomas.
No obstante, para que se produzca una enfermedad, de las llamadas raras, tienen que juntarse los padres con los mismos genes alterados.
De lo contrario, nada raro se desarrollará en nuestro organismo, porque uno de los padres trasmite una copia sana de genes al organismo.
Síntomas de la Fenilcetonuria
En términos generales, los pacientes que sufren de fenilcetonuria nacen con un color de piel muy claro, al igual que su cabello y ojos, ya que en el organismo falta la fenilalanina, que es la responsable de la pigmentación.
Aunque los bebés no presentan síntomas en sus primeros meses de nacido, al poco tiempo lo desarrollan con signos muy peculiares como:
- Erupciones cutáneas.
- Convulsiones.
- Retraso en el desarrollo del bebé.
- Microcefalia.
- Retardo mental.
- Hiperactividad.
- Olor a humedad en su aliento, orina y otros fluidos.
Diagnóstico de la Fenilcetonuria
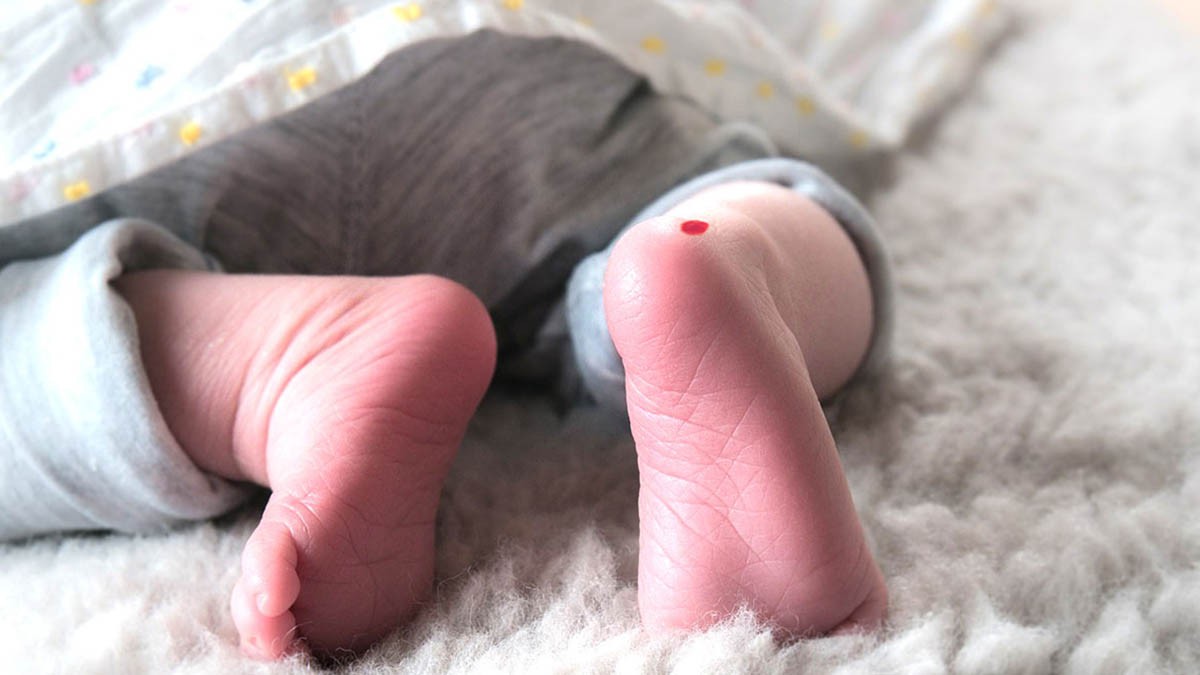
Con una prueba en sangre es suficiente para saber si un bebé sufrirá de fenilcetonuria. Esta debe realizarse después de 24 horas de nacido el bebé, para dar tiempo de que consuma alguna proteína natural o artificial.
Con la prueba se medirá el nivel de proteína en sangre. Si resulta positivo, se procederá a realizar pruebas de orina y pruebas genéticas.
La prueba consiste en un pinchazo en el talón del bebé para extraerle unas gotas de sangre. Esto no acarrea consecuencias mayores, solo llanto y una pequeña marca en su piel.
La prueba es importante, porque a partir de los resultados, se pueden evitar daños severos en el cerebro y mejorar su calidad de vida.
Lo primero que hay que hacer frente a un resultado positivo es organizar una dieta especial que evite los niveles altos de consumo de proteína y la consecuente toxina que afecta el sistema neurológico.
Cuando se tiene un paciente con fenilcetonuria, es importante diagnosticar al resto de la familia. A los padres se les debe diagnosticar para confirmar la alteración genética del bebé.
También es importante que los hermanos sanos se sometan a esta prueba, para conocer su potencial trasmisible y a los familiares cercanos, de acuerdo a su historial médico.
1-Tipos de Fenilcetonuria
Según la gravedad, la fenilcetonuria se clasifica en:
- Severa: En este caso, la enzima que descompone la fenilalanina es inexistente o muy disminuida, lo que acarreará graves daños al cerebro, más si no se diagnostica a tiempo.
- Moderada: La enzima está presente en sangre con niveles moderados, pero es obligatorio mantener una dieta especial que para evitar alteraciones.
2-Fenilcetonuria en el Embarazo
Las mujeres que padecen de fenilcetonuria, y están embarazadas, no pueden dejar de mantener su dieta estrictica. De lo contrario, sufrirán de abortos espontáneos o alterar severamente el desarrollo del feto.
Si la fenilcetonuria es grave o modera, las complicaciones y consecuencias son muy desfavorables para el feto, e inclusive para la madre.
Los bebés nacidos de madres con fenilcetonuria bajo cuidados estrictos, aún pueden nacer bajos de peso, con anomalías faciales, defectos cardíacos, etc.
Cuando se conoce que alguno de los padres es portador del gen alterado, puede aplicarse una prueba al líquido amniótico a las 16 semanas de embarazo, para conocer si el feto será sano, portador o afecto.
Tratamiento de la Fenilcetonuria

El primer tratamiento para la fenilcetonuria es el alimentario y se debe mantener durante toda la vida, porque la metabolización está obstruida y no se debe elevar el consumo de proteínas.
Alimentos que se deben evitar:
- §Carnes: pollo, ternera, cerdo y otros.
- §Embutidos: salchichas, pates y otros.
- §Pescados mariscos.
- §Huevos.
- §Lácteos.
- §Frutos secos: pistachos, avellanas, nueces y otros.
- §Alimentos con gluten (pastas, arroz, harinas).
- §Evitar el espartato.
- §Soja.
¿Qué puede Consumir la Persona con Fenilcetonuria?

Se recomienda una dieta vegetariana estricta. Entre los alimentos a consumir destacan:
- Leche descremada o desnatada.
- Aceite de oliva.
- Setas.
- Tomates.
- Berenjenas.
- Calabacines.
- Yogur natural.
- Maicena, tapioca.
- Azúcar, miel, mermeladas, siropes, caramelos.
En el año 2009, la comunidad médica presentó un fármaco para controlar la fenilalanina, este fue aprobado por la comunidad europea y está disponible en algunos países.
Supone un gran avance que permitirá liberar a las personas de la dieta estricta que debe seguir durante toda la vida.
Dieta PKU
En el período de lactancia, los niños deben tener una dieta exenta de Phe, e iniciarlos prontamente en el consumo de papillas a base de frutas y cereales sin gluten. Las madres que deseen darle lactancia materna sus hijos, deben consultar con su médico.
En la medida que crezca, es posible incorporar alimentos bajos en proteínas o sustitutos de la misma en dosis controladas. Este régimen será durante toda la vida.
Riesgos de la Fenilcetonuria
Los principales riesgos proceden de dos fuentes. La primera tiene que ver con un diagnóstico tardío por no aplicar la prueba al bebé recién nacido u otra causa.
Por otro lado, a consecuencia de esto, la ingesta de alimentos ricos en proteína acelerará el deterioro cerebral, conductual y vital de la persona.
Finalmente, cuando la enfermedad está declara y el paciente no acata la dieta, puede causarse daños irreversibles.